2018年7月16日,程斯进等人的研究论文“Systematic identification and annotation of multiple-variant compound effects at transcription factor binding sites in human genome”于Journal of Genetics and Genomics (JGG)在线发表。该研究中,本课题组针对调控区转录因子结合位点,开发了首个考虑转录因子结合位点序列上下文的变异注释工具COPE-TFBS,并将其应用到千人基因组以及GTEx项目变异数据,鉴定被以往算法忽略的由多个变异引起的复合效应。该工具也是本课题组继COPE-PCG (Cheng et al., 2017)后,另一个基于基因组上下文准确解释调控区基因组变异造成影响的工具,为后续深入探究基于基因组变异解释及预测个体表型鉴定了基础。
转录因子可以通过结合DNA序列(转录因子结合位点,TFBS)调控下游基因的表达(Latchman, 1997)。发生在TFBS内的基因组变异可以通过影响转录因子与DNA的结合强度进而影响正常的生理功能(Huang et al., 2014; Liu et al., 2017)。目前虽然有若干针对TFBS内变异的注释工具(Boyle et al., 2012; Coetzee et al., 2015; Fu et al., 2014; Kumar et al., 2017; Ward and Kellis, 2016; Zuo et al., 2015),但它们都没有考虑变异所在的基因组上下文,因而都不能正确处理多个变异相互作用产生的多变异复合效应。
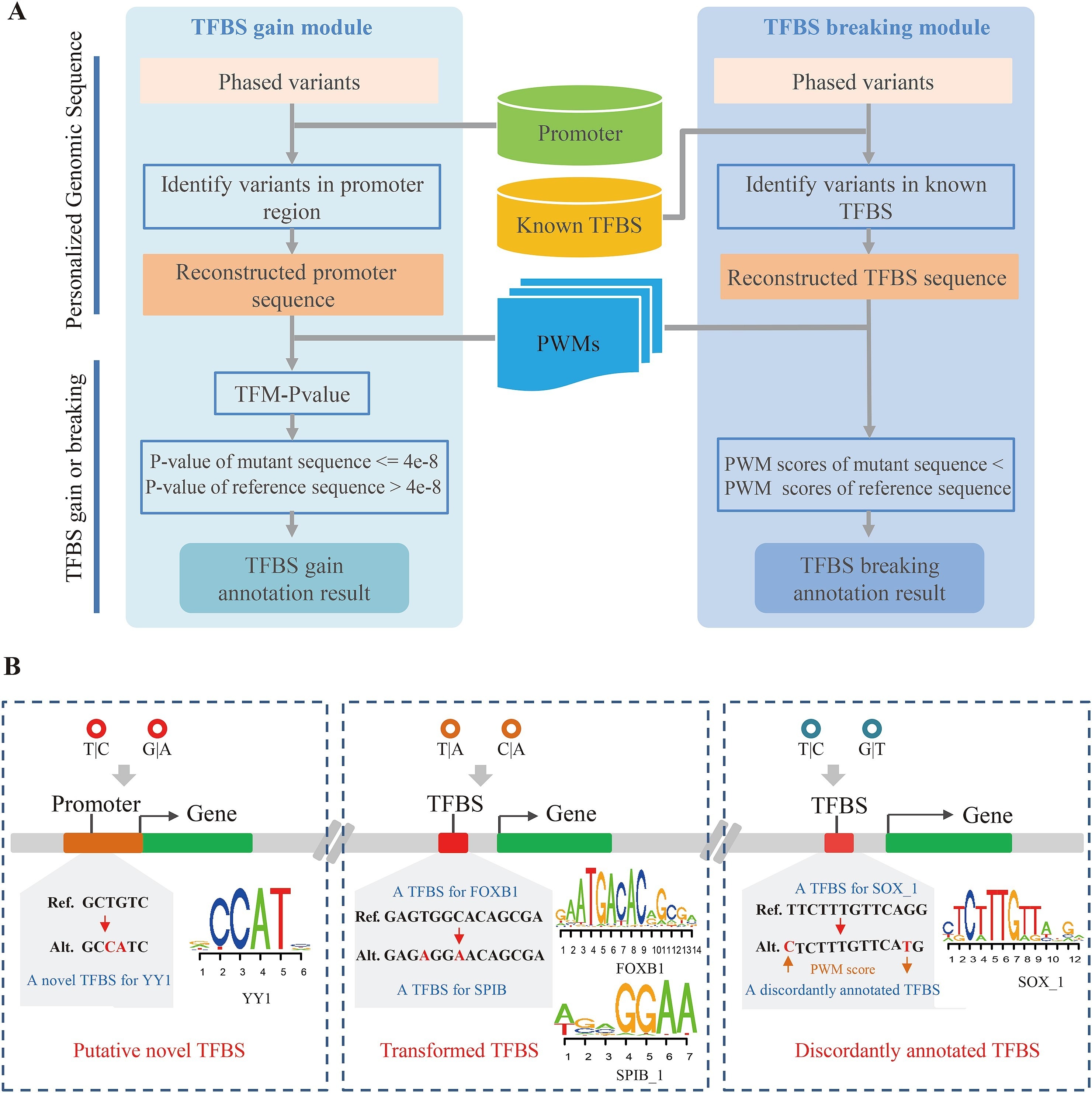
(A) COPE-TFBS的流程图。(B) 发生在TFBS内的多变异复合效应。
为了正确处理发生在TFBS内的多变异复合效应,本研究通过构造变异后的转录因子结合位点序列,计算其位置权重矩阵(position weight matrix, PWM)分值,并与参考序列进行比较,,以此判断基因组变异对TFBS的影响。通过对上千组个人基因组数据进行分析,本研究首次大规模鉴定出1502 个由多个变异共同引入的转录因子结合位点、266 个由多个变异造成的转录因子 结合位点转换事件(从一个转录因子结合位点转换为另一个转录因子结合位点)以及 超过 85000 个由多个变异造成注释不一致的转录因子结合位点(一个变异可以增强转录因子的结合强度,而另一个变异可以减弱转录因子的结合强度),并发现这些复合效应能够影响 eQTL、下游基因的表达以及疾病相关的调控变异。
本研究开发的COPE-TFBS是首个可以正确处理发生在TFBS内的多变异复合效应的变异注释工具,相关服务器目前已公开上线。
原文链接:https://doi.org/10.1016/j.jgg.2018.05.005
参考文献:
Cheng, S.J., Shi, F.Y., Liu, H., Ding, Y., Jiang, S., Liang, N., Gao, G., 2017. Accurately annotate compound effects of genetic variants using a context-sensitive framework. Nucleic acids research 45, e82.
Latchman, D.S., 1997. Transcription factors: an overview. Int J Biochem Cell Biol 29, 1305-1312.
Huang, Q., Whitington, T., Gao, P., Lindberg, J.F., Yang, Y., Sun, J., Vaisanen, M.R., Szulkin, R., Annala, M., Yan, J., Egevad, L.A., Zhang, K., Lin, R., Jolma, A., Nykter, M., Manninen, A., Wiklund, F., Vaarala, M.H., Visakorpi, T., Xu, J., Taipale, J., Wei, G.H., 2014. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nature genetics 46, 126-135.
Liu, N.Q., Ter Huurne, M., Nguyen, L.N., Peng, T., Wang, S.Y., Studd, J.B., Joshi, O., Ongen, H., Bramsen, J.B., Yan, J., Andersen, C.L., Taipale, J., Dermitzakis, E.T., Houlston, R.S., Hubner, N.C., Stunnenberg, H.G., 2017. The non-coding variant rs1800734 enhances DCLK3 expression through long-range interaction and promotes colorectal cancer progression. Nature communications 8, 14418.
Boyle, A.P., Hong, E.L., Hariharan, M., Cheng, Y., Schaub, M.A., Kasowski, M., Karczewski, K.J., Park, J., Hitz, B.C., Weng, S., Cherry, J.M., Snyder, M., 2012. Annotation of functional variation in personal genomes using RegulomeDB. Genome research 22, 1790-1797.
Coetzee, S.G., Coetzee, G.A., Hazelett, D.J., 2015. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 31, 3847-3849.
Fu, Y., Liu, Z., Lou, S., Bedford, J., Mu, X.J., Yip, K.Y., Khurana, E., Gerstein, M., 2014. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome biology 15, 480.
Kumar, S., Ambrosini, G., Bucher, P., 2017. SNP2TFBS – a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic acids research 45, D139-D144.
Ward, L.D., Kellis, M., 2016. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic acids research 44, D877-881.
Zuo, C., Shin, S., Keles, S., 2015. atSNP: transcription factor binding affinity testing for regulatory SNP detection. Bioinformatics 31, 3353-3355.