细胞是生物体生命活动的基本单元,生命活动的正常进行依赖于精确的基因表达调控过程,而基因转录调控过程则是基因表达调控中的重要环节。人类基因组中约98%的区域为非编码区,其中约80%的区域可能涉及基因转录调控过程。基因的时空特异性表达依赖于复杂的基因调控网络,涉及多种顺式调控元件,并且往往以组合的方式共同发挥调控作用。针对高等真核生物多层次、跨尺度的复杂基因调控模式,如何有效鉴定基因组中的调控元件并挖掘转录调控规律是亟待解决的重要挑战。
针对上述挑战,2024年12月30日,北京大学/昌平实验室高歌课题组于Nature Communication在线发表题为Quantifying the regulatory potential of genetic variants via a hybrid sequence-oriented model with SVEN的研究论文,通过引入多尺度混合模型,逐层学习序列中的调控信息,进而量化建模特定调控元件及其组合对下游基因表达的影响。在此基础上,构建了人类转录调控相关非编码变异功能解析方法SVEN,能够针对不同类型的变异(包括大尺度的结构变异和小尺度的SNV/indel等),在超过350个组织和细胞系中实现复杂非编码基因组变异的功能预测与机制解析,为从序列水平深入理解细胞调控图谱提供了有价值的方法学基础与数据储备。

为充分利用现有数据并提升模型的准确性,SVEN采用了一种不同于传统“单一大模型”的构建方式,创新性地引入了基于序列的多尺度混合模型架构:基于来自ENCODE的超过400种组织和细胞系的组学数据,通过结合针对特征的个体模型(feature-oriented separate models)和针对类的整体模型(class-oriented holistic models),以一组“小模型”分别学习基因组调控序列的特征(如转录因子结合、DNA开放性和组蛋白修饰),而后再通过隐空间投影去除冗余的特征信息,实现了对多个组织和细胞系中基因转录水平的量化建模(图1)。
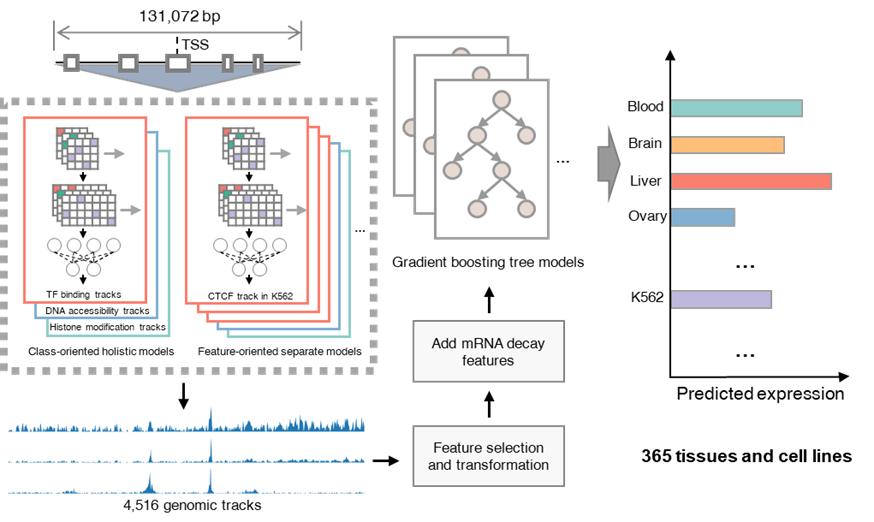
图1 SVEN模型的结构示意图
得益于SVEN的多尺度混合模型的设计,尽管其模型参数更少(SVEN中最大的模型参数量:153M;Enformer:249M),SVEN可以更为准确预测基因在不同组织和细胞系中基因转录水平(图2)。
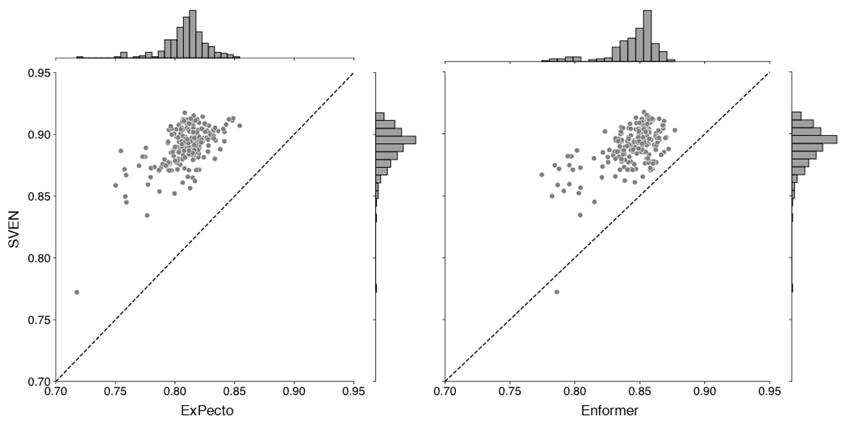
图2 SVEN可准确预测不同组织和细胞系中基因转录水平
基因组变异是指基因组核苷酸序列的改变,其中发生在基因组非编码区的变异被称为非编码变异。全基因组关联研究(GWAS)表明,超过90%与人类疾病或性状相关的变异为非编码变异。大规模人群全基因组测序,绘制了人类基因组变异的高分辨率图谱,涵盖了小尺度变异以及大尺度结构变异(>50bp)。研究表明,结构变异在多种疾病的发生中起着重要作用。然而,在全基因组范围内系统研究结构变异对基因转录调控的影响仍然面临巨大挑战。
得益于创新的模型设计,SVEN能够精确预测结构变异对基因转录水平的影响(图3a)。例如,基因FOLH1编码前列腺特异性膜抗原(prostate-specific membrane antigen,PSMA),其水平升高被认为是前列腺癌和胰腺癌重要的诊断与筛查指标。在基因FOLH1上游存在长为313bp的删除,影响了该基因的启动子区域。传统基于功能注释的方法预测该变异对基因转录水平几乎没有影响(score = -0.02)。然而,SVEN预测该变异会显著提升基因FOLH1的转录水平(log2 fold change = 1.076,A375细胞系),这一结果得到了CRISPR实验的验证(A375细胞系,图3b和3c)。此外,SVEN注释模型显示,该变异导致基因FOLH1转录起始位点附近区域的H3K4me3和H3K27ac激活信号强度提升(图3d),这可能是基因FOLH1转录水平提高的部分原因。
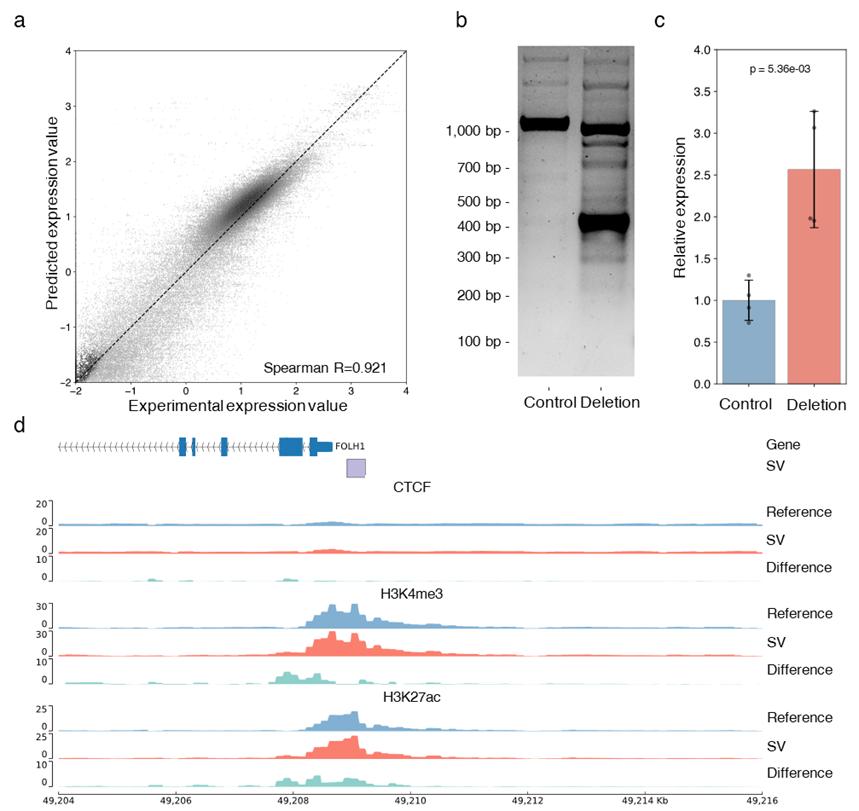
图3 SVEN可准确预测结构变异对基因转录水平影响
SVEN全部实现代码已经开源发布:https://github.com/gao-lab/SVEN。
北京大学生命科学学院博士生王宇(已毕业,现为昌平实验室博士后)为该论文第一作者,梁楠在实验验证方面提供大力支持。该研究得到了国家重点研发计划、蛋白质与植物基因研究国家重点实验室、北京未来基因诊断高精尖创新中心和昌平实验室的资助。计算分析工作于昌平实验室高性能计算平台、北京大学太平洋高性能计算平台与北京大学高性能计算校级公共平台完成。
论文链接: